
Le differenze tra i cinghiali italiani. I più autoctoni sono quelli di Castelporziano, Maremma e Sardegna
I cinghiali italiani altamente differenziati da tutte le altre popolazioni di cinghiale europee analizzate
[28 Febbraio 2022]
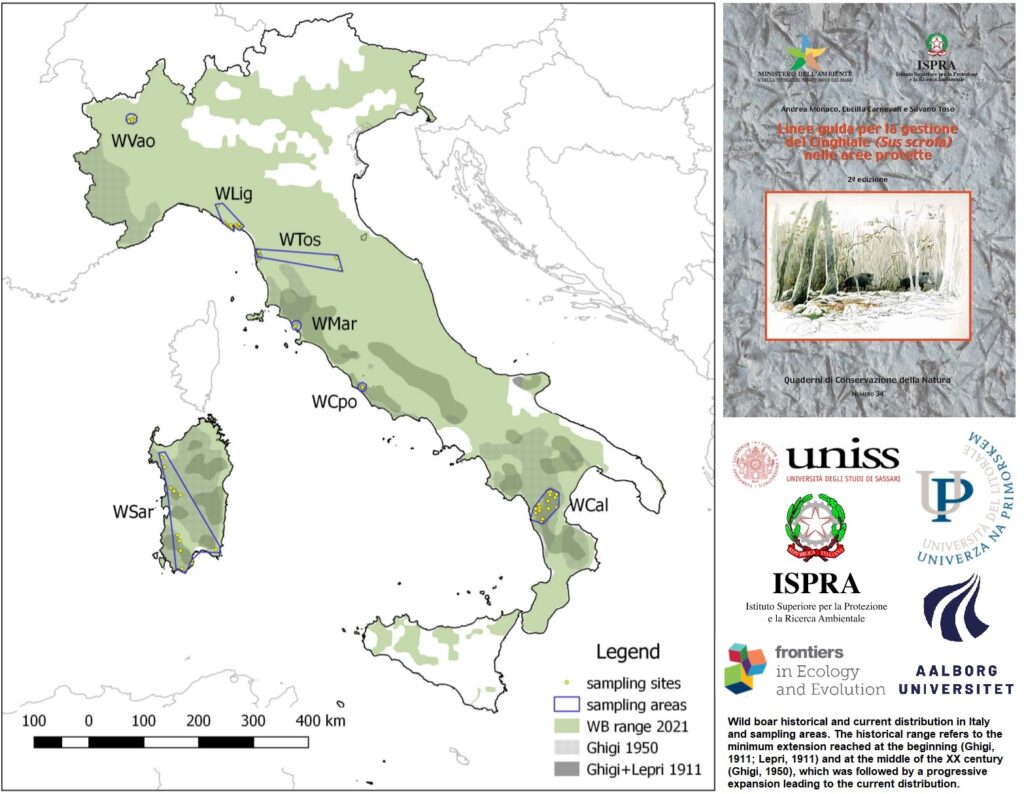
Le attività antropiche possono modificare globalmente gli ecosistemi naturali determinando perturbazioni ecologiche, demografiche e di areale per diverse specie animali. Questi cambiamenti possono mettere a repentaglio in diversi modi i pool genetici autoctoni, portando sia all’omogeneizzazione genetica o, al contrario, alla scissione in demi geneticamente divergenti. Negli ultimi decenni, la maggior parte delle popolazioni di cinghiali europei (Sus scrofa) sono state pesantemente gestite dall’uomo e queste manipolazioni antropiche hanno colpito fortemente anche le popolazioni italiane con la caccia, traslocazioni e reintroduzioni che potrebbero aver modificato fortemente i loro pool genici originari. Lo studio “Resilience to Historical Human Manipulations in the Genomic Variation of Italian Wild Boar Populations”, pubblicato su Frontiers in Ecology and Evolution da Massimo Scandura, Giulia Fabbri, Giulio Pante e Marco Apollonio del Dipartimento di medicina veterinaria dell’università di Sassari, Romolo Caniglia, Federica Mattucci, Chiara Mengoni e Nadia Mucci dell’Area per la Genetica della Conservazione dell’Istituto superiore per la protezione e la ricerca ambientale (BIO–CGE Ispra) e Laura Iacolina dell’Univerza na Primorskem/Università del litorale di Koper, Slovenia e dell’Aalborg Universitet, Aalborg, Danimarca si occupa proprio di questo.
I ricercatori spiegano che «In questo studio, sfruttando la disponibilità di genoma suino ben mappato, abbiamo applicato strumenti genomici per esplorare la variabilità dell’intero genoma nelle popolazioni di cinghiali italiane, indagare sulla loro struttura genetica e rilevare le firme di una possibile introgressione da suini domestici e cinghiali non autoctoni . I dati genomici di 134 cinghiali campionati in 6 aree dell’Italia peninsulare e in Sardegna sono stati raccolti utilizzando il BeadChip Illumina Porcine SNP60 (60k Single Nucleotide Polymorphisms – SNPs) e confrontati con genotipi di riferimento provenienti da esemplari europei e da suini domestici».
La popolazione di cinghiale è un buon esempio dei cambiamenti nelle popolazioni di grandi mammiferi europei: dopo un forte calo che ha portato a una distribuzione frammentata e dispersa, ora l’areale del cinghiale si estende quasi ininterrottamente in tutto il continente fino ai Paesi Baltici, con un trend in costante aumento. In Italia la specie ha avuto un declino demografico e una contrazioni di areale fino alla prima metà del XX secolo, seguito da una continua riespansione che continua ancora e che è favorita da cambiamenti socio-economici che hanno portano al recupero dell’habitat e dal rilascio di individui allevati in cattività o importati.
Lo studio ricorda che «Dopo un progressivo incremento, il cinghiale rappresenta oggi uno degli animali più invasivi e impattanti del Paese, probabilmente anche per il suo corredo genetico che potrebbe aver giocato un ruolo cruciale nel suo adattamento ai diversi contesti ambientali ed ecologici».
Durante la maggiore contrazione del suo areale, il cinghiale italiano era sopravvissuto solo in Maremma, in alcune aree centro-meridionali della penisola, dove nella tenuta presidenziale di Castelporziano persiste una popolazione isolata relitta, e in Sardegna. Nel 1927 De Baux e Festa, nello studio “La Ricomparsa del cinghiale nell’Italia settentrionale-occidentale” pubblicato dalla Società italiana di scienze naturali, dopo aver realizzato analisi morfometriche, proposero che le popolazioni maremmane e sarde fossero classificate come due differenti sottospecie endemiche: Sus scrofa majori e Sus scrofa meridionalis. Ma la ripresa della popolazione di cinghiali che fece seguito alla fine della seconda guerra mondiale, avvenne sia per diffusione naturale delle popolazioni esistenti (come la ricolonizzazione delle Alpi occidentali dalla Francia) sia con processi artificiali come le reintroduzioni e il ripopolamento a fini venatori. che hanno comportato un massiccio rilascio di cinghiali da diverse fonti, che probabilmente si sono mescolati con gli stock selvatici locali.
Nella valutazione “The Eurasian wild pig, Sus scrofa” del 1993, il Gruppo di specialistici dell’IUCN evidenziava che «La popolazione di Castelporziano rappresenta probabilmente l’unico ceppo autoctono di razza pura sopravvissuto in Italia».
I primi studi genetici sul cinghiale italiano, basati sul sequenziamento del DNA mitocondriale (mtDNA), hanno rivelato l’esistenza di una linea endemica del mtDNA italiano, presente sia in campioni storici che moderni, che è finito per essere isolato in Italia durante i periodi glaciali. Ulteriori studi hanno confermato la presenza di variazioni genetiche autoctone anche nelle regioni autosomiche, pur rilevando firme di commistione locale con ceppi non autoctoni. Lo studio fa notare che «Tuttavia, queste informazioni erano solo parziali e non conclusive, poiché questi studi erano basati su un numero limitato di marcatori molecolari e il numero di popolazioni campionate e di individui analizzati era scarsamente rappresentativo della varietà di situazioni che è probabile che si verifichino nella penisola italiana. Dettagli più precisi sono stati ottenuti sulle popolazioni sarde, che, nonostante l’intricato modello di struttura genetica e l’introgressione locale da cinghiali non autoctoni e suini domestici, conservano ancora una proporzione significativa di diversità endemica, distinguendole bene dai relativi conspecifici peninsulari».
Ma tutti gli studi condotti finora sul cinghiale in Italia hanno incontrato le stesse difficoltà nel districare gli effetti locali della manipolazione umana, inclusa la commistione tra individui provenienti da molteplici fonti selvatiche e domestiche. Il nuovo studio, ha esplorato, per la prima volta, la diversità genomica del cinghiale italiano e i ricercatori dicono: «Ci aspettavamo che gli attuali livelli complessivi di diversità e la composizione genomica delle popolazioni italiane fossero largamente influenzati dalle loro storie demografiche e manipolazioni umane. Il progressivo declino e contrazione dell’areale della specie in Italia avvenuta tra la fine del XIX e l’inizio del XX secolo ne provocò la frammentazione in piccoli demi isolati. Questi nuclei sono durati per decenni fino alla più recente riespansione, iniziata dopo la seconda guerra mondiale, e hanno probabilmente subito colli di bottiglia accompagnati da una perdita di variazione genetica e cambiamenti nelle frequenze alleliche. Questo processo storico potrebbe spiegare perché i livelli di diversità genomica sono più bassi in Italia rispetto alla maggior parte delle popolazioni europee, in particolare quelle registrate in aree che hanno subito un isolamento prolungato e forte (es. Tenuta Presidenziale di Castelporziano). Tuttavia, le traslocazioni di animali selvatici e il rilascio di esemplari in cattività avvenuti negli ultimi 60 anni, sono avvenuti senza omogeneità in tutto il Paese e hanno determinato un flusso genico mediato dall’uomo che può essere responsabile di una variazione abbastanza maggiore in alcuni zone, dove individui di diversa origine possono essersi confusi.
Infatti, per molto tempo la legislazione italiana ha consentito il rilascio di cinghiale per il ripopolamento o la reintroduzione. Eppure, dalla fine degli anni ’80 è stato introdotto un divieto, prima nelle singole regioni e poi, solo nel 2015, a livello nazionale».
La popolazione più divergente è risultata essere quella della Tenuta Presidenziale di Castelporziano e «Una divergenza così pronunciata di questa popolazione può essere il risultato di un isolamento duraturo e di dimensioni effettive limitate, che probabilmente hanno modellato le sue frequenze alleliche per deriva genetica». Un’interpretazione simile può essere data per il cinghiale del Parco Regionale della Maremma che ha mostrato un certo livello di divergenza dalle altre popolazioni italiane, ma inferiore a quello di Castelporziano. Nello specifico, «I campioni di Castelporziano hanno mostrato esclusivamente aplotipi del mtDNA appartenenti al ceppo italiano E2, che sono stati rilevati anche in esemplari museali di cinghiale maremmano. La divergenza osservata tra cinghiale du Castelporziano e della Maremma potrebbe quindi, in linea di principio, essere determinata anche da un diverso livello di introgressione rispetto ad altre popolazioni (appenniniche). La selezione artificiale per “morfotipi autoctoni” che è stata effettuata in passato nel Parco Naturale della Maremma potrebbe aver contribuito alla minore introgressione degli individui campionati in quest’area, mentre l’isolamento del cinghiale di Castelporziano avrebbe dovuto prevenire qualsiasi contaminazione genetica»
Tutte le analisi hanno confermato l’elevata differenziazione genomica del cinghiale sardo da tutte le popolazioni della terraferma.
La popolazione di cinghiali valdostana mostra chiaramente una diversa composizione rispetto agli altri campioni italiani e simile alle popolazioni dell’Europa centro-occidentale che si riflette anche nella popolazione piemontese. I ricercatori sottolineano che « Questo risultato, tuttavia, contrasta con il presunto rilascio di ceppi ibridi in cattività nella regione. I nostri dati genomici suggeriscono quindi che i processi naturali (cioè l’espansione dalle aree circostanti) hanno prevalso sulle cause mediate dall’uomo (cioè i rilasci)».
Il team italiano evidenzia che «Un eccezionale risultato inaspettato del nostro studio è stato rappresentato dalla discrepanza tra distanza genetica e spaziale che abbiamo riscontrato. I cinghiali delle popolazioni WTos, WLig e WCal [Toscana, Liguria e Calabria] hanno infatti mostrato un’elevata somiglianza in tutte le analisi condotte, nonostante la loro posizione geografica. È probabile che queste popolazioni rappresentino le popolazioni di cinghiale più manipolate nel nostro campione. La loro composizione genomica potrebbe essere influenzata da una serie di interventi umani (reintroduzioni, traslocazioni) che hanno confuso e diffuso un pool genetico misto, che appare come un cluster separato nelle analisi ADMIXTURE».
All’interno della popolazione di cinghiali della Toscana occidentale i ricercatori hanno riscontrato una diversa composizione genetica tra le località: «Un gruppo di individui campionati nella tenuta di San Rossore, sulla costa tirrenica, si discostava leggermente dagli altri individui WTos della Toscana orientale e anche dalla vicina popolazione WLig, mostrando somiglianze con individui di WMar (Marche). Secondo la sua storia, questo nucleo costiero ha probabilmente una proporzione di variazione nativa maggiore rispetto agli altri individui campionati nella Toscana settentrionale».
Comunque, anche se l’intervento umano spiega i modelli di variazione genomica in Italia osservati dai ricercatori, «I segnali rilevati di mescolanza con maiali domestici cinghiali non italiani nelle popolazioni campionate erano inferiori al previsto sulla base delle conoscenze pregresse. Sia l’analisi ADMIXTURE che quella f3 non hanno mostrato segnali rilevanti di introgressione da fonti esterne nelle popolazioni italiane, in contrasto con i modelli emersi da altri studi basati su analisi di microsatelliti e MC1R».
Per i ricercatori si potrebbero proporre due possibili spiegazioni per i loro risultati: «(1) l’introgressione da più popolazioni europee di cinghiali e maiali era rilevante ma si è verificata molte generazioni fa ed è stata seguita da una divergenza locale del pool genetico misto da tutte le popolazioni parentali; (2) i rilasci di WB di origine straniera o di ibridi cinghiale-maiale sono avvenuti localmente ma hanno coinvolto un numero limitato di individui, che sono stati superati in numero da individui traslocati di origine italiana e/o da popolazioni autoctone in espansione naturale (quindi gli alleli non nativi sono stati diluiti dagli autoctoni). Nella prima situazione, il nuovo pool genetico apparirebbe come un cluster esclusivo diverso nelle analisi ADMIXTURE, in quanto non sarebbe possibile risalire al contributo delle popolazioni parentali. Nel secondo caso, la prevalente origine autoctona degli individui spiegherebbe la constatata mancanza di introgressione; tuttavia, l’origine per traslocazione da più aree in Italia genererebbe comunque un pool genetico misto e unico. Le informazioni storiche disponibili per le nostre aree di campionamento e i risultati delle analisi f3 supportano la seconda linea di interpretazione, considerando l’espansione delle popolazioni autoctone residue sostenute dal ripopolamento umano con cinghiali nativi, come principali fattori di recupero del cinghiale nella penisola. Sia le traslocazioni passate che quelle più recenti infatti, sebbene in parte non documentate, sono state effettuate utilizzando animali ruspanti o in cattività dell’Italia centrale, che derivavano in gran parte dalle popolazioni tirreniche (come la tenuta di Castelporziano o la Maremma)».
Il basso tasso di introgressione da maiale domestico rilevato nel nuovo studio è coerente con i risultati precedenti. Già nel 2018 uno studio che utilizzava lo stesso dataset non aveva rilevato nessun ibrido in un sottocampione di 19 cinghiali in Toscana, mentre 3 ibridi su 25 individui (12%) sono stati rilevati in un campione casuale di cinghiale sardo. Al contrario, utilizzando un approccio a marcatori multipli (colore del mantello, MC1R, SNPs), nel 2022 è stata trovata una sovrapposizione completa tra i genomi degli ibridi conosciuti dell’Italia meridionale con quelli del cinghiale simpatrico, probabilmente come risultato di una profonda introgressione in quest’ultimo.
Ma nel 2016 in Sardegna le analisi f3 hanno rilevato una introduzione di cinghiali continentali, supportando così un flusso genico recente. Anche uno studio del 2011 aveva rilevato una recente introgressione in Sardegna di cinghiali dall’Italia continentale, oltre che da maiali domestici,, mentre l’introgressione reciproca con maiali sardi era stata dimostrata nel 2016 all’analisi della variazione allelica di geni nucleari.
Nonostante l’opinione prevalente tra cacciatori e gestori abbia descritto il cinghiale nativo italiano come quasi estinto geneticamente, il nuovo studio rivela che «Le popolazioni italiane possono ancora mantenere un’elevata percentuale di diversità genomica nativa. La forte divergenza che le popolazioni italiane hanno mostrato da tutti gli altri cinghiali europei e dai maiali domestici, così come la presenza di un lignaggio esclusivo del mtDNA, sono chiari indicatori di una variazione endemica mantenuta.
I ricercatori sono convinti che «I nostri risultati suggeriscono che l’impatto dei processi naturali a lungo termine (come le fluttuazioni climatiche quaternarie, l’espansione dell’areale, la selezione naturale), nonché il prolungato isolamento geografico dell’Italia determinato dalle Alpi e dal Mar Mediterraneo, potrebbero ancora influenzare la struttura filogeografica di un certo numero di specie, prevalendo sugli effetti delle manipolazioni umane nel plasmare i modelli di diversità genetica del cinghiale. Risultati simili sono stati trovati di recente in uno studio genomico su popolazioni gestite di pernici rosse (Alectoris rufa, Forcina et al., 2021)».
Le popolazioni italiane di cinghiali meglio conservate sembrano quelle della costa tirrenica centrale (Castelporziano e Maremma) e «L’elevata differenziazione genomica di queste popolazioni WB, unitamente alla loro unicità morfometrica, richiederebbe una rivalutazione della sottospecie Sus scrofa majori, come già proposto di recente per altre popolazioni di mammiferi italiani altamente differenziati, come il cervo della Mesola (Cervus elaphus italicus), il capriolo italiano (Capreolus capreolus italicus), e il lupo appenninico (Canis lupus italicus)».
Ma sono gli stessi ricercatori autori dello studio a ricirdare che «Tuttavia, le popolazioni di cinghiale italiane che abbiamo studiato, pur contribuendo alla variazione genetica complessiva del cinghiale italiano, rappresentano solo una parte di esso. Le popolazioni dell’Italia nord-orientale, non incluse nel presente studio, hanno mostrato una composizione diversa da quelle che vivono nell’Italia centrale, rivelando maggiori somiglianze con le popolazioni dell’Europa orientale. Allo stesso modo, nell’Italia meridionale, un tasso di introgressione altamente rilevabile da maiale domestico è stato riscontrato nella popolazione di cinghiale che vive a nord fino alla popolazione WCal (area del Cilento), dove è stata anche descritta un’elevata variazione morfologica. Sfortunatamente, nessuna informazione genetica o fenotipica è invece disponibile per il cinghiale reintrodotto che si sta diffondendo in tutta la Sicilia, così come per altre aree di ricolonizzazione più recente. E’ quindi evidente che la popolazione italiana di cinghiale rappresenta un insieme fortemente eterogeneo di demi con storie e composizione genomica differenti, determinato da una combinazione di fattori mediati dall’uomo quali reintroduzioni, traslocazioni, attività di ripopolamento con individui allevati (a volte ibridi), frammentazione dell’intervallo ed eventi demografici, risultando in un’eterogeneità genomica molto più elevata che in qualsiasi altra regione europea».
I ricercatori fanno notare che i dati emersi dallo studio «Riconoscono chiaramente il valore endemico della diversità genomica nativa italiana di cinghiale. Pertanto i nostri risultati ne raccomandano vivamente la conservazione (a) prevenendo un’ulteriore mescolanza tra le popolazioni di cinghiali, regolamentando rigorosamente l’allevamento di cinghiali e vietando permanentemente l’importazione di cinghiali (come attualmente regolamentato per motivi sanitari, cioè la prevenzione della peste suina africana), nonché (b ) riducendo al minimo l’introgressione domestica prevenendo accuratamente l’interazione con le razze suine, soprattutto se allevate allo stato seminaturale, e con gli stock di cinghiale in cattività liberati, possibilmente introgressi deliberatamente da suini domestici».
I ricercatori concludono: «Studi futuri basati sul sequenziamento dell’intero genoma potrebbero contribuire a chiarire definitivamente le storie evolutive e i tempi di divergenza delle popolazioni di cinghiale italiane attraverso l’analisi delle traiettorie demografiche e dei modelli di flusso genico passato con altre popolazioni selvatiche e razze domestiche europee. Gli approcci paleogenomici e del DNA antico potrebbero inoltre aiutare a valutare quanta parte dell’attuale diversità negli stock locali di cinghiale può essere attribuita a un pool genetico nativo confrontando campioni moderni e storici (es. esemplari museali o trofei impagliati) datati prima del 1950, quindi prima della maggior parte del flusso genico mediato dagli umani. Inoltre, le analisi dell’intero genoma e del trascrittoma potrebbero aiutare a identificare ulteriori evidenze di differenziazione genetica e variazione adattativa nel cinghiale italiano, che non è stato possibile rilevare dal chip Porcine SNP60 a causa di limitazioni nel suo sviluppo (esclusi gli animali italiani). Tali ulteriori regioni genomiche potrebbero consentire di indagare possibili modelli di selezione o adattativi nelle popolazioni WB italiane, ospitanti potenzialmente geni responsabili del loro adattamento locale all’ambiente mediterraneo e, teoricamente, per la loro elevata divergenza fenotipica. Infine, la disponibilità di interi genomi potrebbe anche far luce sul potenziale evolutivo a lungo termine delle popolazioni italiane di cinghiale, sulla loro capacità di affrontare attivamente i cambiamenti climatici in atto e sul loro futuro ruolo ecologico nell’Antropocene».